In my last post, I argued that a major distinction in machine learning is between predictive learning and representation learning. Now I'll take a stab at summarizing what representation learning is about. Or, at least, what I think of as the first principal component of representation learning.
A good starting point is the notion of representation from David Marr's classic book, Vision: a Computational Investigation [1, p. 20-24]. In Marr's view, a representation is a formal system which "makes explicit certain entities and types of information," and which can be operated on by an algorithm in order to achieve some information processing goal (such as obtaining a depth image from two stereo views). Representations differ in terms of what information they make explicit and in terms of what algorithms they support. He gives the example of Arabic and Roman numerals (which Shamim also mentioned): the fact that operations can be applied to particular columns of Arabic numerals in meaningful ways allows for simple and efficient algorithms for addition and multiplication.
I'm sure a lot of others have proposed similar views, but being from MIT, I'm supposed to attribute everything to Marr.
I'm not going to say much about the first half of the definition, about making information explicit. While Marr tends to focus on clean representations where elements of the representation directly correspond to meaningful things in the world, in machine learning we're happy to work with messier representations. Since we've built up a powerful toolbox of statistical algorithms, it suffices to work with representations which merely correlate enough with properties of the world to be useful.
What I want to focus on is the second part, the notion that representations are meant to be operated on by algorithms. An algorithm is a systematic way of repeatedly applying mathematical operations to a representation in order to achieve some computational goal. Asking what algorithms a representation supports, therefore, is a matter of asking what mathematical operations can be meaningfully applied to it.
Consider some of the most widely used unsupervised learning techniques and the sorts of representations they produce:
- Clustering maps data points to a discrete set (say, the integers) where the only meaningful operation is equality.
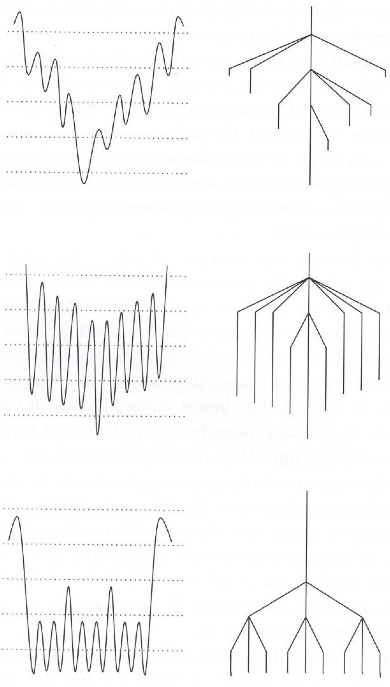
- Nonlinear dimensionality reduction algorithms (e.g. multidimensional scaling, Isomap) map data points to a low-dimensional space where Euclidean distance is meaningful.
- Linear dimensionality reduction algorithms like PCA and factor analysis map data points to a low-dimensional space where Euclidean distance, linear combination, and dot products are all meaningful.
This list doesn't tell the whole story: there are a lot of variants on each of these categories, and there are more nuances in terms of precisely which operations are meaningful. For instance, in some versions of MDS, only the rank order of the Euclidean distances is meaningful. Still, I think it gives a useful coarse description of each of these categories.
There are also a wide variety of representation learning algorithms built around mathematical operations other than the ones given above:
- Greater/less than. TrueSkill is a model of people's performance in multiplayer games which represents each player with a scalar, and whoever has a larger value is expected to win. [2]
- $L_p$ distance. There is a variant of MDS which uses general $L_p$ distance rather than Euclidean distance. [3]
- Hamming distance. Semantic hashing is a technique in image retrieval which tries to represent images in terms of binary representations where the Hamming distance reflects the semantic dissimilarity between the images. The idea is that we can use the binary representation as a "hash key" and find semantically similar images by flipping random bits and finding the corresponding hash table entries. [4]
- Projection onto an axis. We might want the individual coordinates of a representation to represent something meaningful. As Shepard discusses, MDS with L1 distance often results in such a representation. Another MDS-based technique he describes is INDSCAL, which tries to model multiple subjects' similarity judgments with MDS in a single coordinate system, but where the individual axes can be rescaled for each subject. [3]
- Matrix multiplication. Paccanaro and Hinton [5] proposed a model called linear relational embedding, where entities are represented by vectors and relations between them are represented by matrices. Matrix multiplication corresponds to function composition: if we compose "father of" and "father of," we get the relation "paternal grandfather of."
Roger Shepard's classic 1980 paper, "Multidimensional scaling, tree fitting, and clustering," [3] highlights a variety of different representation learning methods, and I took a lot of my examples from it. The paper is written from a cognitive science perspective, where the algorithms are used to model human similarity judgments and reaction time data, with the goal of understanding what our internal mental representations might be like. But the same kinds of models are widely used today, albeit in a more modern form, in statistics and machine learning.
One of the most exciting threads of representation learning in recent years has been learning feature representations which could be fed into standard machine learning (usually supervised learning) algorithms. Depending on the intended learning algorithm, the representation has to support some set of operations. For instance,
- For nonparametric regression/classification methods like nearest neighbors or an SVM with an RBF kernel, we want Euclidean distance to be meaningful.
- For linear classifiers, we want dot products to be meaningful.
- For decision trees or L1 regularization, we want projection onto an axis to be meaningful.
For instance, PCA is often used as a preprocessing step in linear regression or kernel SVMs, because it produces a low-dimensional representation in which Euclidean distance and dot products are likely to be more semantically meaningful than the same operations in the original space. However, it would probably not be a good preprocessing step for decision trees or L1 regularization, since the axes themselves (other than possibly the first few) don't have any special meaning. The issues have become somewhat blurred with the advent of deep learning, since we can argue endlessly about which parts of the representation are semantically meaningful. Still, I think these issues at least implicitly guide the research in the area.
The question of which operations and algorithms are supported is, of course, only one aspect of the representation learning problem. We also want representations which where both the mapping and the inverse mapping can be computed efficiently, which can be learned in a data efficient way, and so on. But I still find this a useful way to think about the relationships between different representation learning algorithms.
[1] David Marr. Vision: a Computational Investigation. W. H. Freeman and company, 1982.
[2] Ralf Herbrich, Tom Minka, and Thore Graepel, TrueSkill(TM): A Bayesian Skill Rating System, in Advances in Neural Information Processing Systems 20, MIT Press, January 2007
[3] Roger Shepard. "Multidimensional scaling, tree-fitting, and clustering." Science, 1980.
[4] Ruslan Salakhutdinov and Geoffrey Hinton. "Semantic Hashing." SIGIR Workshop on Information Retrieval and Applications of Graphical Models, 2007.
[5] Alberto Paccanaro and Geoffrey Hinton. "Learning distributed representations of concepts using linear relational embedding." KDE 2000.